Cancer Institute NSW Early Career Fellow and Senior Data Scientist at CMRI, and Adjunct Lecturer at the University of Sydney. I build deep-learning methods — federated, generative, and transformer-based — for cancer multi-omics, aimed at translation into clinical precision oncology.
Zhaoxiang Cai, Emma L Boys, Zaynab Noor, …, Roger R Reddel
The proteome provides unique insights into disease biology beyond the genome and transcriptome. However, the sharing of raw proteomic data across institutions is hindered by privacy concerns and data volume. Here, we present a federated deep learning framework for cancer subtyping using mass spectrometry-based proteomic data. By training on distributed datasets without centralized data sharing, our approach achieves performance comparable to centralized training. We demonstrate the utility of this framework by classifying 14 cancer subtypes across 7,500 cancer proteomes from multiple centers. This work introduces the first application of federated deep learning to cancer proteomics, enabling collaborative research while preserving data privacy.
Zhaoxiang Cai, Rebecca C Poulos, Jianmin Liu, Qing Zhong
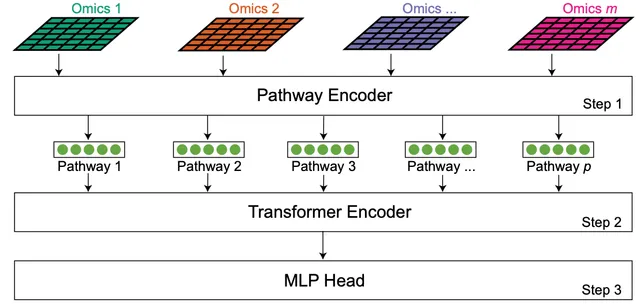
DeePathNet is a transformer-based deep learning model that integrates multi-omic data with biological pathway information. By embedding pathway knowledge directly into the model architecture, DeePathNet improves the interpretability and performance of cancer subtype classification and drug response prediction. We demonstrate the utility of DeePathNet on large-scale datasets, highlighting its ability to identify pathway-level biomarkers and mechanisms of action.
Zhaoxiang Cai, Samuel Apolinário, Ana R Baião, …, Emanuel Gonçalves
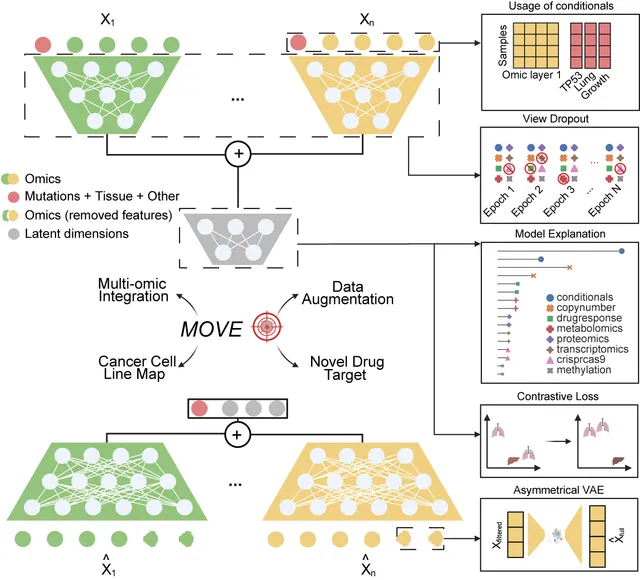
We introduce MOSA (Multi-Omic Synthetic Augmentation), an unsupervised deep learning model for integrating and augmenting cancer multi-omics data. By leveraging variational autoencoders, MOSA generates synthetic multi-omic profiles that expand the effective sample size of cancer datasets, enabling the discovery of new biomarkers and drug targets. We demonstrate that MOSA-augmented data improves the power of association studies and clustering analyses, providing a valuable resource for the cancer research community.
Zhaoxiang Cai, Emanuel Gonçalves, Rebecca C Poulos, …, Roger R Reddel
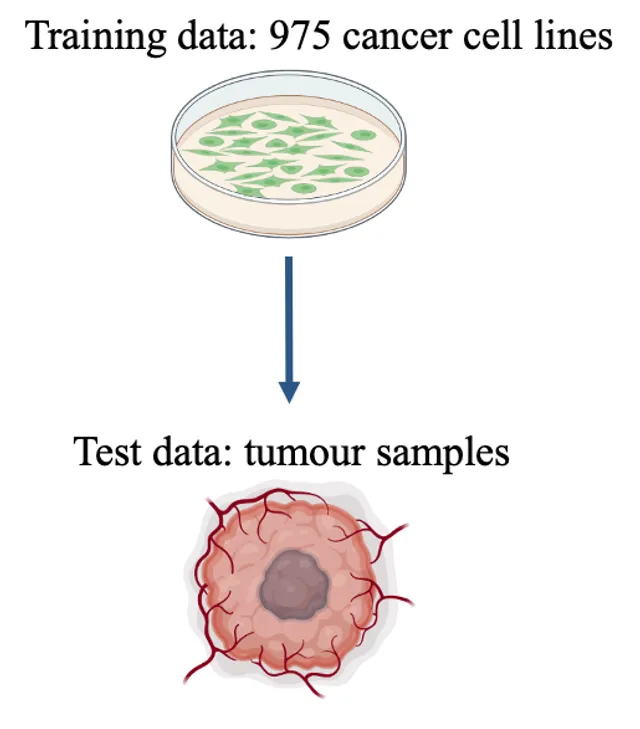
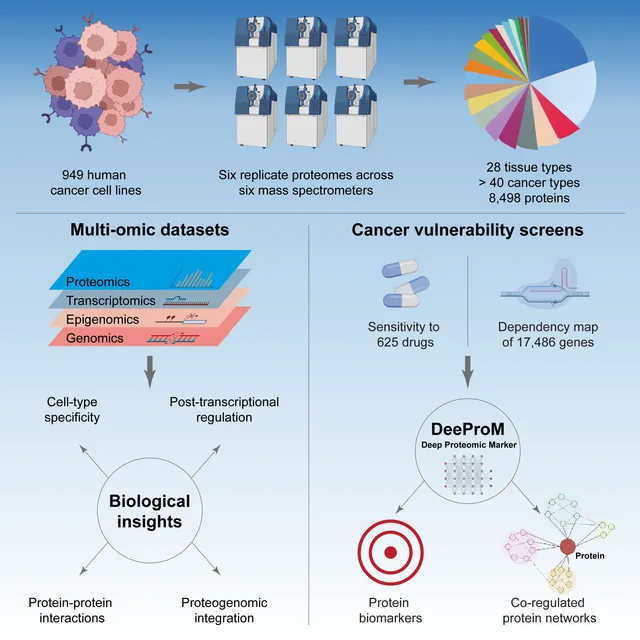
The proteome provides unique insights into disease biology beyond the genome and transcriptome. A lack of large proteomic datasets has restricted the identification of new cancer biomarkers. Here, proteomes of 949 cancer cell lines across 28 tissue types are analyzed by mass spectrometry. Deploying a workflow to quantify 8,498 proteins, these data capture evidence of cell-type and post-transcriptional modifications. Integrating multi-omics, drug response, and CRISPR-Cas9 gene essentiality screens with a deep learning-based pipeline reveals thousands of protein biomarkers of cancer vulnerabilities that are not significant at the transcript level. The power of the proteome to predict drug response is very similar to that of the transcriptome. Further, random downsampling to only 1,500 proteins has limited impact on predictive power, consistent with protein networks being highly connected and co-regulated. This pan-cancer proteomic map (ProCan-DepMapSanger) is a comprehensive resource available at https://cellmodelpassports.sanger.ac.uk.
The sharing of sensitive patient data across institutions is a major bottleneck in cancer research. To address this, we developed a federated deep learning framework that enables collaborative training of cancer subtyping models without sharing raw data. By keeping data local and only sharing model updates, we can leverage the collective power of distributed datasets while preserving patient privacy. Our results demonstrate that federated models achieve performance comparable to centralized training, paving the way for large-scale, multi-institutional collaborations in precision oncology. This project culminated in the first application of federated learning to cancer proteomics, published in *Cancer Discovery*.
Multi-omic data analysis incorporating machine learning has the potential to significantly improve cancer diagnosis and prognosis. Traditional machine learning methods are usually limited to omic measurements, omitting existing domain knowledge, such as the biological networks that link molecular entities in various omic data types. Here we develop a Transformer-based explainable deep learning model, DeePathNet, which integrates cancer-specific pathway information into multi-omic data analysis. Using a variety of big datasets, including ProCan-DepMapSanger, CCLE, and TCGA, we demonstrate and validate that DeePathNet outperforms traditional methods for predicting drug response and classifying cancer type and subtype. Combining biomedical knowledge and state-of-the-art deep learning methods, DeePathNet enables biomarker discovery at the pathway level, maximizing the power of data-driven approaches to cancer research.
Integrative analysis of multi-omic datasets remains a challenge due to gaps and heterogeneity. We present a bespoke unsupervised deep learning model that generates synthetic multi-omic data for 1,523 cancer cell lines, completing the gaps and increasing the number of molecular and phenotypic profiles by 32.7%. Our model augments cellular measurements, improves cancer type clustering, and increases statistical power for cancer dependency biomarker discovery. Model explanation facilitates biomarker discovery and cancer target prioritization.
Deep learningMulti-view Variational AutoencoderMulti-omic integration
Cancer type is determined via assessment of tumour morphology, aided by immunohistochemical staining patterns. The development of machine learning (ML) models using histology slides has powered the image-based prediction of the site of origin in cancer of unknown primary (CUP). Here, we present an ML-based method to predict cancer type from a pan-cancer cohort consisting of 1,289 human tissue samples spanning 44 cancer types and 26 different tissues based on proteomic data. All samples were processed using data-independent acquisition mass spectrometry (DIA-MS). Two proteomic profiles from the pan-cancer cell line cohort were generated using two different sample preparation methods. These were normalized and merged by averaging the protein abundance, yielding a single training set (D1) with 975 cell lines and 9,688 proteins. Similarly, 1,277 tissue samples were processed by DIA-MS, quantifying 9,501 proteins. We trained a classifier using the cell lines (D1) as the baseline training set, and consecutively added 10% of D2 to D1 for online ML. We tested the baseline model and each subsequent new model on the test set T1. We observed a monotonic performance increase from 0.89 (baseline; Top-1 accuracy) to 0.97 (all D2 were used) when predicting the six cancer types. We observed an analogous trend when predicting the seven tissue types (from 0.64 to 0.84). Our proteomic-based ML model can predict cancer type and carcinoma tissue of origin in concordance with existing histopathological classification. It can also assign multiple probabilities to tumour type and tissue of origin, potentially enabling the classification of challenging pathology cases, such as CUP in future work. By adding tissue samples stepwise to the existing model, its predictive performance can be further enhanced. This reflects a real-world knowledge base that will continue to increase in predictive power with additional incremental proteomic data..
Proteomic data can reveal novel associations between genotype and phenotype, beyond what is apparent from genomics or transcriptomics alone. However, a lack of large proteomic datasets across a range of cancer types has limited our understanding of proteome network organisation and regulation. We produced a pan-cancer proteomic map derived from 949 human cancer cell lines. The map encompasses more than 40 cancer types derived from over 28 distinct human tissues. The samples were processed with a clinically-relevant workflow involving rapid and minimally complex sample preparation, quantifying 8,500 proteins. The raw proteomic data were acquired by data independent acquisition mass spectrometry (DIA-MS) at ProCan® in Australia. The processed data were analysed with a bespoke deep learning-based pipeline (DeeProM) that integrates multi-omics, CRISPR-Cas9 gene essentiality and drug sensitivity information produced at the Wellcome Sanger Institute. First, our findings reveal pervasive post-transcriptional modification and thousands of putative protein biomarkers of cancer vulnerabilities. Second, DeeProM statistics show that a fraction of the proteome can confer similar predictive power to the entire transcriptome. This has key implications for the clinical application of proteomics in drug response prediction. Third, we demonstrate that a random proportion of the identified proteins can provide robust predictions of cancer cell phenotypes, underpinning the concept of pervasive co-regulation of protein networks. This pan-cancer cell line proteomic map is a comprehensive resource that expands our understanding of cancer proteomes. These data reveal principles of cancer cell phenotypes, including genetic vulnerabilities and drug sensitivities, that are important for developing novel targeted anticancer therapies.