Pan-cancer proteomic map of 949 human cell lines reveals principles of cancer vulnerabilities
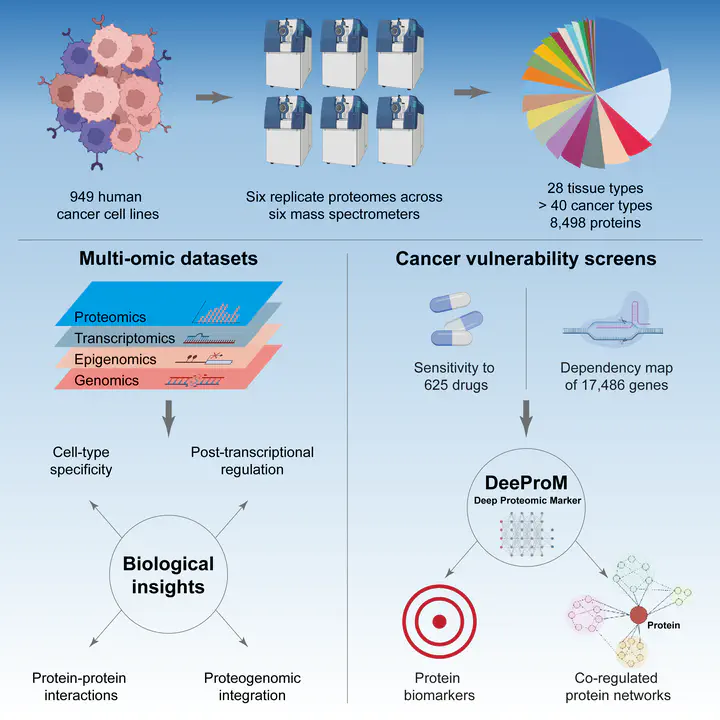
Proteomic data can reveal novel associations between genotype and phenotype, beyond what is apparent from genomics or transcriptomics alone. However, a lack of large proteomic datasets across a range of cancer types has limited our understanding of proteome network organisation and regulation. We produced a pan-cancer proteomic map derived from 949 human cancer cell lines. The map encompasses more than 40 cancer types derived from over 28 distinct human tissues. The samples were processed with a clinically-relevant workflow involving rapid and minimally complex sample preparation, quantifying 8,500 proteins. The raw proteomic data were acquired by data independent acquisition mass spectrometry (DIA-MS) at ProCan® in Australia. The processed data were analysed with a bespoke deep learning-based pipeline (DeeProM) that integrates multi-omics, CRISPR-Cas9 gene essentiality and drug sensitivity information produced at the Wellcome Sanger Institute. First, our findings reveal pervasive post-transcriptional modification and thousands of putative protein biomarkers of cancer vulnerabilities. Second, DeeProM statistics show that a fraction of the proteome can confer similar predictive power to the entire transcriptome. This has key implications for the clinical application of proteomics in drug response prediction. Third, we demonstrate that a random proportion of the identified proteins can provide robust predictions of cancer cell phenotypes, underpinning the concept of pervasive co-regulation of protein networks. This pan-cancer cell line proteomic map is a comprehensive resource that expands our understanding of cancer proteomes. These data reveal principles of cancer cell phenotypes, including genetic vulnerabilities and drug sensitivities, that are important for developing novel targeted anticancer therapies.
Zhaoxiang (Simon) Cai
Senior Data Scientist
Innovative researcher and engineer with experience in both academia and industry.